| Главная » Статьи » Химия » Химия |
C, N, O-ацилированиеC-, N-,O- ацилирование в органической химии Ацилирование - введение ацильной группы (ацила) RCO в молекулу органического соединения путем замещения атома водорода. В широком смысле ацилирование это замещение любого атома или группы атомов на ацила. В зависимости от атома к которому присоединяют ацил различают C-, N-, O-, S- ацилирование… (Химическая энциклопедия, том I, стр. 233). Реакции ацилирования находят широкое применение в современной органическом синтезе. Наиболее распространенными являются реакции С‑, O‑ и N‑ацилирования, с помощью которых получают соответственно кетоны, сложных эфиры или амиды. Частными случаями реакции ацилирования являются реакции формилирования (введение НСО‑группы), ацетилирования (СН3СО‑группы) и бензоилирования (С6Н5СО‑группы). Несмотря на различия в методах проведения реакций, общим для всех вариантов ацилирования является реакция некого субстрата и ацилирующего агента, протекающая как правило по механизму электрофильного замещения. Наиболее распространенными ацилирующими агентами являются ангидриды и хлорангидриды карбоновых кислот. Реакции С‑ацилирования Наиболее распространенной реакцией С-ацилирования является открытая еще в XIX веке реакция Фриделя‑Крафтса. Механизм этой реакции достаточно долго оставался загадкой, однако теперь является точно установленным, что реакция протекает по механизму электрофильного замещения. Реакции электрофильного замещения в ароматическом ряду протекают по одному и тому же пути и начинаются с атаки электрофильной частицы (будь то катион или положительно заряженный конец сильнополяризованной связи) на ароматическую p-электронную систему. При этом образуется резонансно стабилизированный неароматический s-комплекс, или бензониевый ион. За этим следуют потеря протона и сопутствующая ей реароматизация с образованием соединения, в котором электрофильная частица заменила атом водорода у первоначально атакованного углерода. В случае реакции ацилирования в качестве ацилирующего агента как правило применяются ацилхлориды в присутствии кислот Льюиса (чаще всего хлорида алюминия). В таком случае реакция начинается с взаимодействия ацилхлорида с катализатором с образованием собственно электрофильного агента:
Положение равновесия зависит от природы реагентов и от растворителя: высокая диэлектрическая проницаемость сдвигает равновесие в сторону образования карбениевого иона. Затем электрофильный агент реагирует с ароматической молекулой:
Выбор катализатора определяется реакционной способностью ароматического соединения. Чаще всего применяют хлорид алюминия и только для очень реакционноспособных систем (например, для тиофена) используются хлорид цинка, серную кислоту и др. Тригалогениды алюминия образуют комплексы и с ацилирующим средством, и с образующимся карбонильным соединением; комплекс с последним в условиях реакции устойчив. Для синтезов по Фриделю-Крафтцу с ацилгалогенидами необходимы поэтому по меньшей мере мольные количества катализатора. При взаимодействии с ангидридами кислот получающаяся кислота связывает еще один моль катализатора, поэтому в целом необходимы по крайней мере два моль катализатора. В каждом случае по окончании реакции образовавшийся комплекс кетона с хлоридом алюминия должен быть гидролитически разрушен (соляной кислотой со льдом). Реакцию ацилирования по Фриделю-Крафтцу удается распространить на ароматические углеводороды (в том числе полициклические), галогенпроизводные, реакционноспособные гетероциклы (например, тиофен, фуран). Ароматические амины образуют с катализатором неацилирующийся комплекс. Если же аминогруппа защищена ацетилированием, то реакция удается. Ароматические соединения с сильноинактивирующими заместителями, например с нитро-, циан- и карбонильными группами, не ацилируются по Фриделю-Крафтцу. Поэтому при ацилировании можно не опасаться вторичного и полизамещения. Интересным случаем реакции Фриделя-Крафтца является взаимодействие с ангидридами дикарбоновых кислот, приводящее к образованию оксокислот, которые в дальнейшем можно перевести в хиноны:
Растворителем при ацилировании по Фриделю-Крафтцу может служить избыток ацилируемого углеводорода. Иногда применяют дисульфид углерода, так как он практически не влияет на реакционную способность хлорида алюминия, но комплекс образовавшегося ароматического кетона с хлоридом алюминия остается при этом чаще всего в твердой фазе, поэтому при больших загрузках реакционная смесь с трудом перемешивается и обрабатывается. Кроме того, дисульфид углерода ядовит и очень легко воспламеняется. В нитробензоле или галогеноуглеродах (дихлорэтане или трихлороэтилене) активность катализатора несколько понижена из-за комплексообразования, кроме того ацилирование по Фриделю-Крафтцу в них можно применять только при температурах ниже 50 °C, так как в противном случаем они сами вступают в реакцию. В менее полярном 1,2-дихлорэтане (этиленхлориде) из нафталина получают a-кетон и, напротив, в сильнополяризованной среде (нитробензола) - b-кетон. Общая методика ацилирования по Фриделю-Крафтцу. В трегорлой колбе емкостью 1л, снабженной мешалкой и капельной воронкой с хлоркальциевой трубкой, смешивают 400мл 1,2-дихлорэтана с 1,2 моля тонкорастертого хлорида алюминия и добавляют по каплям при перемешивании и охлаждении ледяной водой 1,05 моля ацилхлорида. Затем из капельной воронки при охлаждении водой добавляют 1 моль ароматического соединения так, чтобы температура смеси поддерживалась около 20°С. Реакционную смесь перемешивают еще 1 ч. и оставляют на ночь. При ацилировании галогенбензолов нагревают 5 ч. при 50°С, причем ацилируемое соединение используемся в качестве растворителя (все количества галогенбензола сразу помещают в колбу). Для разложения комплекса кетона с хлоридом алюминия содержимое колбы осторожно выливают на 500мл льда, выпадающий гидроксид алюминия переводят в раствор, добавляя небольшое количество концентрированной соляной кислоты. Затем в делительной воронке отделяют органический слой, а водные дважды извлекают дихлорэтаном. Объединенные вытяжки тщательно промывают водой, 2%-ным раствором гидроксида натрия и снова водой. После сушки карбонатом калия растворитель отгоняют, а кетон перегоняют в вакууме. Приведенная методика также пригодна для полумикросинтезов. Реакции О‑ацилирования. Реакция этерификации, являющаяся по сути реакцией О‑ацилирования, используется в качестве основного способа получения сложных эфиров. Вследствие малой активности карбонильных групп в карбоновых кислотах они, как правило, медленно реагируют со спиртами. Этерификацию можно существенно ускорить, добавляя сильные кислоты (серную кислоту, безводный хлороводород, сульфоновые кислоты, кислые ионообменные смолы):
Скорость этерификации карбоновой кислоты, как и следовало ожидать, возрастает вместе с ростом положительного заряда на карбонильном углероде, т.е. с ростом кислотности. Так, муравьиная, щавелевая, пировиноградная кислоты реагируют достаточно быстро и без добавления катализатора. Сильно влияют на этерификацию стерические факторы. С ростом объема алкильных остатков, связанных с карбоксильной группой, а также спиртовым гидроксилом, скорость этерификации падает. Поэтому разветвление у a-углеродного атома, а также о-замещенные ароматические кислоты вступают в реакцию медленно и с плохими выходами. В ряду от первичных к третичным спиртам реакция затрудняется еще и тем, что в условиях реакции (сильнокислая среда) параллельно возрастает и тенденция к превращению спиртов в простые эфиры и олефины. Эфиры третичных спиртов получаются прямой этерификацией лишь с очень малыми выходами. Равновесие реакции этерификации не очень благоприятно для получения сложных эфиров. Равновесие можно сдвинуть вправо, используя 5-10-кратный избыток более дешевого исходного вещества (им обычно бывает спирт) либо постоянно удаляя из реакционной смеси продукты реакции - воду или сложный эфир. Общие методики этерификации карбоновых кислот. I. Связывание воды водоотнимающими средствами. Смешивают 1 моль карбоновой кислоты (или 0.5 моля дикарбоновой), 5 молей соответствующего абсолютного спирта и 0.2 моля концентрированной серной кислоты и кипятят без доступа воздуха с обратным холодильником 5 ч. В случае менее стойких вторичных спиртов лучше не применять в качестве катализатора серную кислоту, а насытить кипящую смесь хлороводородом и увеличить продолжительность кипячения до 10 ч. После этого отгоняют главную массу избыточного спирта на колонке Вигре длинной 20 см и выливают остаток в пятикратный объем ледяной воды. Органический слой отделяют, а водный раствор нейтрализуют водным трижды экстрагируют эфиром. Объединенные органические слои нейтрализуют раствором карбоната натрия, промывают водой до нейтральной реакции, сушат хлоридом кальция и перегоняют. Синтез может быть проеден и в полумикромасштабе. II. Азеотропная этерификация Смешивают 1 моль карбоновой кислоты (или 0.5 моль дикарбоновой), 1.75 моля спирта (можно и не абсолютного), 5 г концентрированной серной, толуолсульфоновой, нафталинсульфоновой кислоты или ионообменной смолы в Н-форме, например вофатита KPS, и 100 мл хлороформа или тетрахлоридуглерода. Смесь кипятят с обратным холодильником и водоотделителем, пока не прекратится выделение воды. При этерификации гидроксикислот, a, b-ненасыщенных кислот, а также при этерификации вторичными спиртами лучше не применять серную кислоту, чтобы исключить побочные процессы. При использовании ионообменной смолы жидкость необходимо перемешивать, в противном случае жидкость может выбрасывать из колбы. При окончании реакции смесь охлаждают, отмывают водой применявшуюся в качестве катализатора кислоту, промывают водным раствором гидрокарбоната натрия, еще раз водой, либо отфильтровывают ионообменную смолу. После этого отгоняют растворитель, который захватывает с собой примесь воды, остаток перекристаллизовывают и перегоняют. Синтез можно проводить и в полумикромасштабе. III. Экстрактивная этерификация. Смешивают 1 моль карбоновой кислоты, 3 моля метанола на каждую карбоксильную группу, 300 мл тетрахлоридуглерода (или 1,2-дихлороэтана, или трихлороэтилена) и 5 мл концентрированной серной кислоты (в случае менее устойчивых веществ берут 5 г толуолсульфоновой кислоты или ионообменной смолы) и кипятят 10 ч с обратным холодильником без доступа влаги воздуха. В случае ароматических карбоновых кислот применяют трехкратное количество катализатора. Обычно образуются два слоя; слой меньшего объема содержит воду. После охлаждения органический слой промывают водой, водным раствором гидрокарбоната и снова водой. Растворитель, служивший для экстракции, отгоняют, а остаток перегоняют или перекристаллизовывают. Для получения сложных эфиров можно использовать также в качестве исходных веществ эфиры соответствующей кислоты с другими спиртами. Подобный алкоголиз эфиров карбоновых кислот (переэтерификация) может в отличии от обычной этерификации катализироваться как кислотами, так и основаниями. В данном случае также имеют место типичные равновесные превращения. Из-за весьма повышенной карбонильной активности алкоголиз ангидридов кислот и ацилхлоридов происходит гораздо легче, чем алкоголиз карбоновых кислот и их сложных эфиров. Однако и в этом случае кислоты и основания оказывают ускоряющее действие на реакцию. В отличии от первичных спиртов эфиры третичных спиртов могут быть получены с помощью ангидридов кислот и ацилгалогенидов. Например, трет-бутиловой эфир уксусной кислоты можно получить из уксусного ангидрида и трет-бутилового спирта в присутствии хлорида цинка. Фенолы не этерифицируются непосредственно карбоновыми кислотами. Сложные эфиры получают действием ангидридов и хлорангидридов карбоновых кислот.
Реакции N‑ацилирования. Реакции N-ацилирования, взаимодействие аммиака или аминов с ацилирующим агентами, приводит к образованию аминов
Образовавшаяся кислота HL связывает эквивалентное количество непрореагировавшего амина. Данный метод становится неэкономичным, если амин трудно синтезировать или он представляет собой дорогостоящий реактив. По этой причине амины часто ацилируют при помощи так называемой реакции Шоттена-Баумана, которая сводится к взаимодействию между амином и ацилирующим агентом в присутствии водного раствора едкого натра. Реакция состоит из двух стадий, в процессе которых амин успешно конкурирует с гидроксид-ионом и ацилируется, а гидроксид-ион связывает образующую кислоту (HCl).
Третичные амины взаимодействуют с хлоангидридами, давая четвертичные соли, которые сами являются ацилирующими агентами. Примером такой реакции служит образование солей в пиридине. такая соль ацилирует воду. Процесс в целом представляет собой гидролиз ацилгалогенида, катализируемый пиридином.
| |
| Просмотров: 3980 | Комментарии: 1 | |
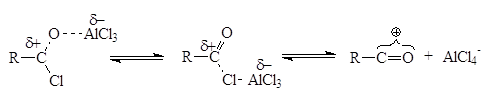
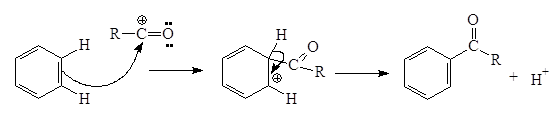
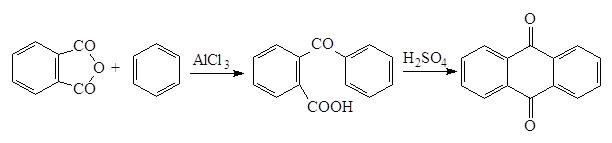
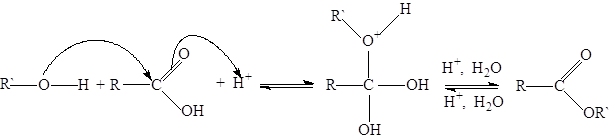
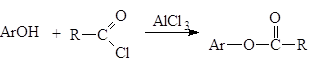
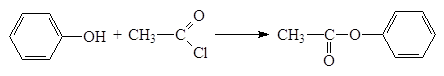
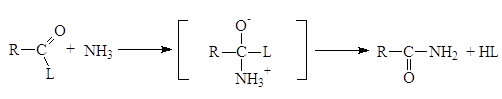
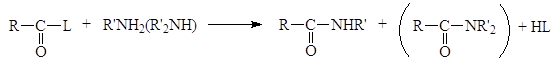
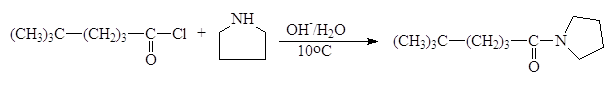
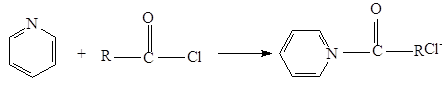

| Всего комментариев: 0 | |
| Химия [30] |
| Золото [4] |
| Химические элементы в организме человека. [6] |
| Менделеев Дмитрий [7] |
| Органическая химия [7] |